Dever ético dos profissionais da saúde
19 de fevereiro de 2009 | Autor: antonini
Ao contrário do que se poderia imaginar, quando um
fármaco novo é introduzido no mercado, dispõe-se, de maneira geral,
apenas de dados suficientes para assegurar que sua margem de segurança é
aceitável e, segundo Laporte e cols., neste momento pouco ainda se
conhece sobre seus efeitos no organismo. Isto ocorre porque os ensaios
clínicos aos quais os fármacos são submetidos antes de entrarem no
mercado são limitados, não sendo capazes de detectar, por exemplo,
reações adversas raras ou associadas ao uso prolongado.Quando um novo
fármaco chega às prateleiras das farmácias, todo o conhecimento que se
tem a seu respeito até então foi extraído dos estudos
pré-comercialização. Nesses estudos, inicialmente avalia-se a toxicidade
da nova molécula em animais de laboratório (estudos ou ensaios
pré-clínicos) e, caso a toxicidade não seja inaceitável, são realizados
estudos em humanos (estudos ou ensaios clínicos) para investigação de
aspectos relacionados com a farmacocinética, a eficácia e a toxicidade.
Mesmo com a realização desses ensaios, os dados obtidos são ainda
considerados limitados, já que existem diferenças importantes entre as
condições de uso do fármaco nos estudos clínicos e na prática clínica
habitual.
Durante os estudos pré-comercialização, um número restrito de pacientes
é utilizado (raramente chega a cada dos milhares), a duração do uso é
relativamente curta (de dias a semanas), geralmente evitam-se outros
tratamentos concomitantes e excluem-se pacientes com contra-indicações
potenciais como gestantes, crianças e idosos. Além disso, os pacientes
são acompanhados de perto por profissionais da área de saúde e recebem
informações sobre o tratamento. Já quando o fármaco chega ao mercado,
seu uso pode atingir dezenas de milhares de pessoas (inclusive
gestantes, crianças e idosos), os tratamentos às vezes duram anos, os
pacientes usam outros fármacos simultaneamente e o seguimento é menos
rigoroso. No que se refere à toxicidade, estas diferenças condicionam
limitações no conhecimento de interações potenciais, efeitos derivados
do uso crônico e das conseqüências do uso do fármaco em uma população
não selecionada. Além disso, os efeitos indesejáveis, especialmente os
graves, são também raros (freqüência entre 0.1 e 0,01%) ou muito raros
(< 0,01%), podendo não serem identificados durante os estudos
pré-comercialização.
Entende-se hoje, portanto, que o monitoramento da segurança dos fármacos
não se encerra quando o produto chega ao mercado, mas sim que neste
ponto é iniciada uma nova fase, chamada de vigilância
pós-comercialização, estudos de fase IV ou farmacovigilância. Este
acompanhamento das informações sobre segurança pode conduzir a melhorias
e mudanças mais rápidas nas informações de registro (por exemplo,
alterações na bula) ou, até mesmo, à retirada do produto do mercado.
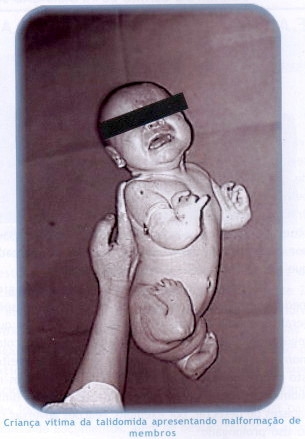
Em 1961, milhares
de crianças nasceram com malformações de membros como conseqüência do
uso do antiemético talidomida por suas mães durante a gravidez. Após
esta tragédia as autoridades governamentais adotaram uma nova postura
diante do risco de produtos farmacológicos, o que culminou
posteriormente com a implantação do Programa Mundial de Monitorização de
Medicamentos, com sede no Centro de Monitorização de Uppsala (The
Uppsala Monitoring Centre), na Suécia. Este centro mantém um banco de
dados internacional de reações adversas de fármacos (RAM) e presta
serviço aos centros nacionais de farmacovigilância associados ao
programa e tem sede na Unidade de Farmacovigilância da ANVISA.
A alimentação dos bancos de dados de RAM é feita principalmente através
do sistema de notificação voluntária (ou espontânea). Neste tipo de
notificação, o notificador (geralmente um profissional de saúde)
preenche uma ficha em que são descritas informações indispensáveis para
que a suspeita seja avaliada e posteriormente a envia para um centro
regional de farmacovigilância. A revisão periódica das notificações
permite identificar associações até então desconhecidas entre fármacos e
RAM, gerando assim um sinal de alerta. Estes sinais são hipóteses sobre
as quais se fazem seguimentos mais intensivos e podem constituir a base
para iniciar estudos epidemiológicos que permitam chegar ao
estabelecimento do risco.
Uma das importantes atribuições que a OMS confere aos Farmacêuticos é a
monitorização dos fármacos. Em muitos casos, quando o paciente apresenta
uma reação adversa, o primeiro profissional que ele procura é um
Farmacêutico na farmácia comunitária. Neste momento o Farmacêutico deve
avaliar a situação relatada pelo usuário coletar informações e enviá-las
para a base de dados regional de farmacovigilância. Muitos Farmacêuticos
ainda não tem o hábito de notificar por razões diversas como receio de
ser penalizado por algo que tenha feito errado, desconhecimento da
importância da notificação ou desinteresse. Para afastarmos receios e
dúvidas é importante ter me mente os seguintes aspectos:
O que deve ser notificado?
Reações adversas, principalmente aquelas consideradas graves (que
acarretam morte ou hospitalização, por exemplo) e raras (como aquelas
que não estão descritas na bula). Não é necessário ter certeza de que se
trata de uma RAM, basta que haja a suspeita. também podem ser
notificadas queixas técnicas e erros de medicação.
Quem deve notificar?
Principalmente os profissionais da saúde (todos, especialmente os
Farmacêuticos). Usuários e empresas farmacêuticas também podem
notificar.
De que forma a notificação pode ser feita?
No Paraná as notificações podem ser feitas através da página da
Secretaria de Estado da Saúde (SESA-PR) na Internet, no endereço
http:://www.saude.pr.gov.br/visa/farmaco/index.html. Escolha o item
Formulário para profissionais de saúde e então imprima o formulário
Reações Adversas e Desvio de Qualidade.
As informações devem ser preenchidas e a ficha
enviada para o fax (41)3330-4543.
